




























Step 3 of 7
Couplage ventriculo-artériel
Une pression est le résultat d'une force contre une résistance. La pression artérielle est le résultat de la mise sous tension et de l’éjection du volume systolique ventriculaire contre les parois artérielles et les embranchements vasculaires périphériques. Elle est le fruit du travail ventriculaire, exprimé par le volume systolique et le flux sanguin, et de la résistance artérielle totale, exprimée par l'impédance aortique, l'onde réfléchie, les RAS et la rigidté ou la souplesse des vaisseaux.
Pression artérielle systémique
La pression artérielle (PA) comprend deux composantes : la pression moyenne (PAM), qui assure un flux distal continu et reste pratiquement identique tout au long de l’arbre artériel (± 2 mmHg), et la pression pulsée (ou pression artérielle différentielle (PP = PAsyst – PAdiast) qui reflète l’intermittence de l’éjection ventriculaire [28]. A partir du calcul des résistances artérielles (RAS = (PAM – PVC) / DC) et sans tenir compte de la pression veineuse, on peut déduire que la pression est le produit des résistances artérielles et du débit cardiaque : PAM = RAS • DC. L’arbre artériel consiste en deux entités anatomiques différentes.
- L’aorte est ses principales branches ont une grande visco-élasticité grâce à leur haute teneur en élastine, ce qui leur permet d’emmagasiner une partie de l’énergie systolique en se dilatant ; cette énergie est restituée en diastole sous forme de pression et de flux ; elles ont une fonction de tampon sur la pulsatilité ventriculaire. Cela a pour effet de diminuer la pression systolique et d’augmenter la pression diastolique (contre-pulsion physiologique). Ce phénomène de tamponnement est essentiel pour rendre le flux quai-continu au niveau capillaire; il disparaît lorsque les vaisseaux se sclérosent et se calcifient avec l’âge et l’athéromatose. Ainsi la pression systolique de la personne âgée s’élève parce que ses vaisseaux sont rigides, mais sa diastolique baisse parce qu’il n’y a plus de restitution de pression en diastole.
- Les artères périphériques agissent comme des conduits de résistance élevée ; elles sont riches en collagène et en musculature lisse. Leurs embranchements sont le lieu de réflexion de l’onde de pression.
Le volume et la pression d’éjection du ventricule sont conditionnés par l’impédance et la résistance exercée par l’arbre vasculaire. Plusieurs phénomènes interviennent lorsque le volume systolique parvient dans les troncs artériels : visco-élasticité de l’aorte, onde réfléchie, impédance et résistance artérielles, compliance artérielle globale, volume éjecté, viscosité (hématocrite). En systole, l’élasticité de l’aorte et des grandes artères emmagasine du volume et de l’énergie en se dilatant. L’impédance, qui se définit comme le rapport entre la pression instantanée et le flux à cet instant (temps t) (Pt / Ft), varie au cours de la systole; elle augmente avec la pression. En diastole, cette énergie est restituée sous forme d’une augmentation de la pression et du flux diastolique. La résistance a lieu dans les artérioles et les capillaires. Elle est définie par la loi d’Ohm, mais ne s’applique qu’à un flux continu non-pulsé. Le volume systolique total est la somme du volume transmis par l'aorte en systole et du volume restitué en diastole (Figure 5.60).
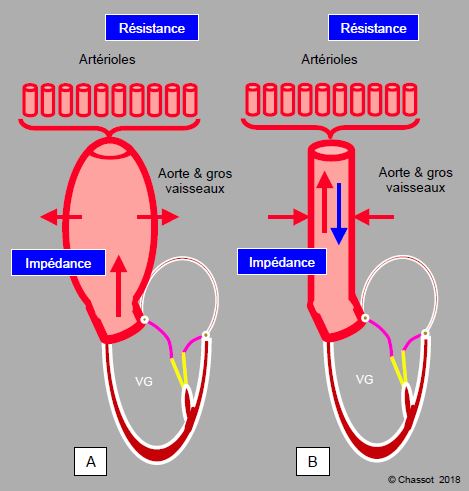
Figure 5.60 : Impédance et résistance artérielles. A : En systole, l’élasticité de l’aorte et des grandes artères emmagasine de l’énergie en se dilatant. L’impédance se définit comme le rapport entre la pression instantanée et le flux à cet instant (temps t) (Pt / Ft) ; elle varie au cours de la systole. B : En diastole, cette énergie est restituée sous forme d’une augmentation de la pression et du flux diastolique. La résistance a lieu dans les artérioles et les capillaires. Elle est définie par la loi d’Ohm, mais ne s’applique qu’à un flux continu non-pulsé. La flèche bleue à contre-courant illustre l’onde de pression rétrograde.
La pression est liée au flux, puisqu'elle augmente lorsque le volume systolique ou la contractilité s'élèvent. Le modèle du Windkessel, élaboré il y a plus d'un siècle, est une tentative de dériver le flux sanguin à partir de la courbe de pression artérielle par analogie à un circuit électrique comprenant trois éléments (Figure 5.61) [5,28].
- Une impédance (Z), représentant la résistance dans un système pulsatile; elle est l'élément essentiel dans les gros vaisseaux, et le facteur principal du stress de paroi maximal pour le VG.
- Une capacitance (C), représentant la compliance artérielle; celle-ci n'est pas linéaire mais décroît avec l'augmentation de pression dans l'aorte.
- Une résistance (R), représentant les résistances artérielles périphériques (RAS) qui sont statiques; la résistance est négligeable dans les vaisseaux de > 5 mm.
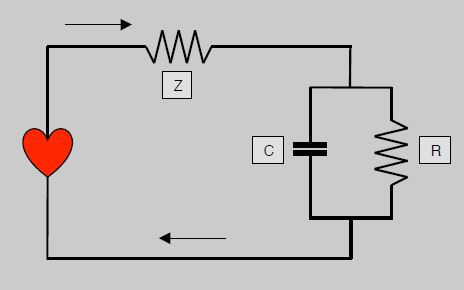
Figure 5.61 : Circuit de Windkessel modélisant l'hémodynamique en circuit électrique. L'impédance (Z) est sur le circuit principal, qui comprend en parallèle une capacitance (C) représentant l'élasticité de l'aorte et une résistance (R) représentant les résistances périphériques.
Dans ces conditions, le flux aortique (Qao) répond à la formule: Qao = (Psyst / Rtot) + Ca (dP/dt), où Psyst est la pression systolique dans l'aorte, Rtot la somme de l'impédance et des RAS, Ca la compliance artérielle et dP/dt le taux d'élévation de la pression par unité de temps. Cette équation est la base de plusieurs dispositifs de monitorage non-invasif du débit cardiaque, mais elle ne tient pas compte du flux lié à la pression réfléchie; de plus, elle suppose que la résistance totale et la compliance artérielle restent constantes au cours du cycle cardiaque, ce qui n'est pas le cas [28].
Pression artérielle réfléchie
La mise sous tension du volume sanguin pendant la contraction isovolumétrique provoque une secousse qui va cheminer dans l’arbre vasculaire sous forme d’une onde de pression, qui ne correspond pas au déplacement du volume éjecté mais le précède (Figure 5.62). Cette onde de pression avance à 3-5 m/s chez le jeune, mais jusqu’à 15 m/s chez le vieillard, car son arbre vasculaire calcifié transmet mieux et plus vite les pressions que les vaisseaux souples du jeune homme [12,13]. Le volume sanguin avance beaucoup moins rapidement : il est éjecté par le VG avec une vélocité de 1.0 - 1.5 m/s, et la vélocité moyenne du flux dans l’aorte est de 0.5 – 1.0 m/s.
L’onde de pression se réfléchit en périphérie lorsque les artères se divisent en fines artérioles riches en collagène mais pauvres en élastine et que les résistances augmentent soudainement; le coefficient de réflexion est de 50-80%; il baisse en cas de vasodilatation et augmente en cas de vasoconstriction [21]. Cette réflexion issue d'innombrables points va renvoyer une onde de pression commune en direction du cœur, pour qui elle représente 10-20% de la postcharge. De plus, elle est associée à un flux rétrograde qui ampute le flux artériel antérograde (Figure 5.63) [22]. Elle parvient normalement au cœur en protodiastole et fonctionne comme une contre-pulsion physiologique, mais sa progression est plus ou moins rapide selon le degré de vasoconstriction et de rigidité de l'arbre artériel. Lorsque celui-ci est rigide, le retour est plus précoce, et l’onde de pression réfléchie vient se superposer en méso-télésystole à la pression systolique engendrée par le volume sanguin éjecté du ventricule. Elle provoque une augmentation de pression par rapport à la pression protosystolique due à l'éjection. Le coefficient d'augmentation est le rapport entre cette valeur d'augmentation (pression systolique maximale – pression d'éjection) et la pression pulsée: Caug = Paug/PP [30].
L’onde de pression se réfléchit en périphérie lorsque les artères se divisent en fines artérioles riches en collagène mais pauvres en élastine et que les résistances augmentent soudainement; le coefficient de réflexion est de 50-80%; il baisse en cas de vasodilatation et augmente en cas de vasoconstriction [21]. Cette réflexion issue d'innombrables points va renvoyer une onde de pression commune en direction du cœur, pour qui elle représente 10-20% de la postcharge. De plus, elle est associée à un flux rétrograde qui ampute le flux artériel antérograde (Figure 5.63) [22]. Elle parvient normalement au cœur en protodiastole et fonctionne comme une contre-pulsion physiologique, mais sa progression est plus ou moins rapide selon le degré de vasoconstriction et de rigidité de l'arbre artériel. Lorsque celui-ci est rigide, le retour est plus précoce, et l’onde de pression réfléchie vient se superposer en méso-télésystole à la pression systolique engendrée par le volume sanguin éjecté du ventricule. Elle provoque une augmentation de pression par rapport à la pression protosystolique due à l'éjection. Le coefficient d'augmentation est le rapport entre cette valeur d'augmentation (pression systolique maximale – pression d'éjection) et la pression pulsée: Caug = Paug/PP [30].
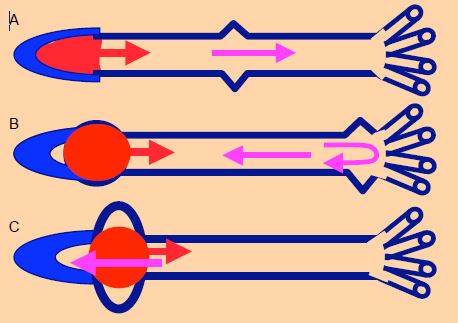
Figure 5.62 : Onde de pression et flux artériel systolique. A : En protosystole, la mise sous tension du volume systolique crée une onde de pression qui se propage dans l’arbre vasculaire en fonction de sa rigidité (flèche violette) ; cette progression est plus rapide en cas d’hypertension ou d’artériosclérose. Elle varie de 4 à 15 m/s. B : L’onde de pression est réfléchie en périphérie au niveau de l’embranchement des artérioles ; elle revient vers le cœur pendant que l’onde de flux commence sa progression (vélocité : 0.5-1.5 m/s). C : L’onde de pression et le bolus systolique se croisent, créant localement une surpression [15]. Les vélocités normales et les distances sont telles que l’onde de pression revient à la racine de l’aorte en protodiastole. Si l’arbre vasculaire est rigide, l’onde de pression chemine plus rapidement et revient à l’aorte en systole ; elle se superpose alors à l’onde de flux sur la courbe artérielle.
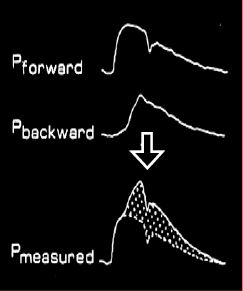
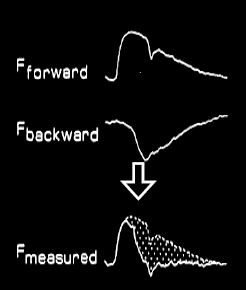
Figure 5.63 : Pression et flux artériels. A gauche, enregistrements décomposés de l’onde de pression antérograde et de l’onde de pression rétrograde dans l’aorte ; leur somme donne la courbe de pression effectivement enregistrée par le cathéter intra-aortique. A droite, enregistrement du flux antérograde lié à la pression intraventriculaire et du flux rétrograde lié à l’onde de pression ; leur somme donne une onde de flux exclusivement systolique dont la forme est très différente de celle de l’onde de pression [Extrait de: Yin FCP, ed. Ventricular/vascular coupling. Clinical, physiological and engineering aspects. New York: Springer Verlag, 1987, Figure 6.6].
La sommation de la pression réfléchie et de la pression systolique augmente la postcharge et le stress de paroi du VG en télésystole proportionnellement à l'amplitude de la première [5]. Toutefois, elle n'est présente que si la fonction ventriculaire gauche est préservée. Lors de défaillance ventriculaire, elle tend à disparaître parce que le flux systolique est trop faible pour compenser le flux rétrograde dû à la réflexion: l'onde réfléchie tronque le flux antérograde, abaisse le volume systolique et raccourcit la durée d'éjection effective [30]. Les vasodilatateurs prescrits dans l'insuffisance cardiaque atténuent la réflexion périphérique et affaiblissent la composante systolique rétrograde; ils améliorent de ce fait le débit antérograde.
La courbe de pression artérielle est différente selon l’endroit de l’arbre vasculaire où elle analysée (Figure 5.64). En avançant vers des zones de moins en moins compliantes, le bolus sanguin rencontre des résistances de plus en plus élevées. Sa synchronisation avec l’onde de pression est différente selon l’endroit où on l'observe. Il est donc normal que les valeurs de la pression systolique (PAs) et de la pression diastolique (PAd) soient différentes selon le point de mesure : la PAs est plus basse dans l’aorte que dans l’artère radiale, alors que la PAd est plus haute. La pression pulsée (PP) augmente en allant vers la périphérie artérielle; seule la pression moyenne (PAM) reste identique [18]. Ce phénomène est accentué lorsque les RAS augmentent.
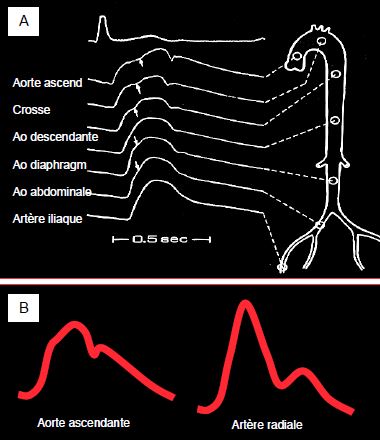
Figure 5.64 : Variations de la forme analogique de la courbe de pression artérielle selon la localisation de l’analyse. A : Image de la courbe de pression normale depuis la racine de l’aorte ascendante jusqu’à l’artère fémorale [Extrait de: Yin FCP, ed. Ventricular/vascular coupling. Clinical, physiological and engineering aspects. New York: Springer Verlag, 1987, Figure 6.5]. B : Image schématique d’une courbe artérielle dans l’aorte ascendante (ponction peropératoire directe) et dans l’artère radiale (cathéter). La pression systolique enregistrée dans la radiale est plus élevée, mais la diastolique est plus basse ; la pression différentielle (Psyst – Pdiast) est agrandie mais la pression moyenne (PAM) est identique.
Un cathéter fémoral lit la pression dans l’artère iliaque externe, qui est un vaisseau élastique ; cette pression est plus proche de celle de l’aorte (donc de celle du cerveau, des reins et des coronaires) que la pression lue dans l’artère radiale, qui est un vaisseau musculaire périphérique soumis aux effets de la vasodilatation ou de la vasoconstriction (Figure 5.65A). Lors d’une choc hypovolémique ou après une CEC en hypothermie profonde, par exemple, il arrive que la vasoconstriction soit telle que la pression lue dans l’artère radiale est la moitié de celle enregistrée dans l’artère fémorale (voir Figure 7.48). Cette dernière est donc un meilleur guide dans les conditions hémodynamiques difficiles.
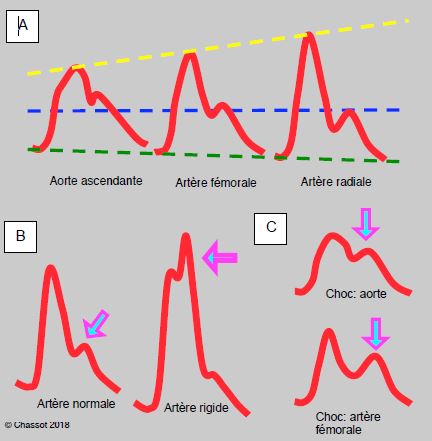
Figure 5.65 : Modifications de forme de la courbe artérielle. A. Image schématique d’une courbe artérielle dans l’aorte ascendante (ponction peropératoire directe) et dans l’artère fémorale ou radiale (cathéter). La pression systolique enregistrée dans la radiale est plus élevée, mais la diastolique est un peu plus basse ; la pression différentielle (Psyst – Pdiast) est agrandie. La pression moyenne (traitillé bleu) est pratiquement identique dans les trois vaisseaux (± 2 mmHg). B. Comparaison d’une artère normale et d’une artère rigidifiée par une athéromatose diffuse (artère fémorale). L’effet du retour de l’onde de pression (onde réfléchie, marquée par la flèche violette) survient plus tôt dans le deuxième cas et donne un crochetage sur le pic systolique. Bien qu’enregistré comme la pression systolique par le moniteur, cet effet ne correspond pas à un flux de perfusion réelle mais à un simple pic de pression. C : Comparaison de la pression dans l’aorte et dans l’artère fémorale en cas de choc hypovolémique. L’onde réfléchie est importante à cause de la vasoconstriction périphérique, mais elle progresse plus lentement à cause de l’hypotension due au faible remplissage (parois vasculaires molles) ; elle est donc très marquée mais décalée dans le temps, d’où l’aspect bifide de la courbe.
L’image de la courbe artérielle sur l’écran du moniteur est très instructive, pour autant que son amplification permette une lecture adéquate. Une artère normale se distingue nettement d’une artère rigide caractérisée par un double pic systolique, typique du patient âgé dont les vaisseaux sont calcifiés (Figure 5.65B) ; dans ce cas, le moniteur enregistre la pression maximale, mais cette dernière correspond souvent à l’onde de pression, non au flux sanguin. Elle ne traduit donc pas une pression de perfusion réelle pour les organes. Dans un état de choc, la vasoconstriction augmente la réflexion de l’onde de pression, mais l’hypotension en diminue la vélocité [19] ; la courbe artérielle apparaît alors bifide dans une artère périphérique (fémorale ou radiale) (Figure 5.65C). C’est une image que l’on constate fréquemment chez les patients en choc hypovolémique.
On voit de ces exemples que l’image analogique de la courbe artérielle offre de nombreux renseignements sur l'hémodynamique et l'état vasculaire du patient. La surface sous la courbe est proportionnelle au volume systolique. La pente ascensionnelle (dP/dt) est le reflet de la performance systolique du ventricule, pour autant qu’il n’y ait pas d’obstacle entre le VG et le cathéter (sténose aortique). La pente télésystolique et la position du dicrotisme sont fonction des RAS (Figure 5.66).
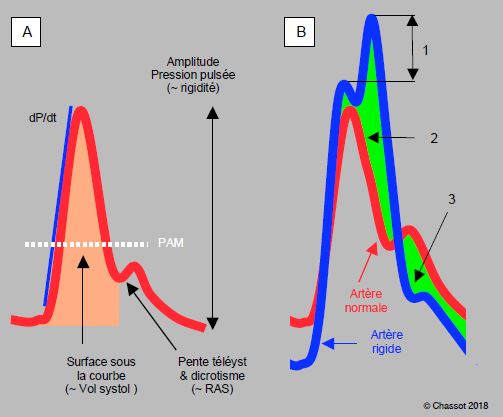
Figure 5.66 : Aspect analogique de la courbe artérielle. A : courbe normale. La pente ascensionnelle est fonction du dP/dt intraventriculaire (pour autant qu’il n’y ait pas de pathologie valvulaire aortique). La surface sous la courbe systolique est proportionnelle au volume systolique, la pente télésystolique et le niveau du dicrotisme sont fonction des résistances artérielles périphériques (RAS), l’amplitude est fonction de la rigidité des parois mais aussi de la volémie et des RAS. La pression artérielle moyenne (PAM) est calculée selon la formule : PAM = (PAsyst + 2 PAdiast)/3. B : comparaison d’une courbe artérielle normale (en rouge) et de la courbe d’un patient souffrant d’athéromatose (en bleu), dont l’aorte est devenue rigide. 1 : augmentation de la pression systolique due à la superposition de l’onde réfléchie. Le coefficient d'augmentation est le rapport entre cette valeur d'augmentation (pression systolique maximale – pression d'éjection) et la pression pulsée. 2 : augmentation de la postcharge du VG. 3 : diminution de la pression de perfusion coronarienne en diastole (PA diastolique plus basse).
Si la surface sous la courbe en systole représente le volume systolique, celle sous la courbe diastolique représente le flux diastolique résultant du volume et de l'énergie emmagasinés dans l'aorte en systole et restitués en diastole. Le flux diastolique est d'autant plus important que la pente de la courbe est plus oblique (Figure 5.67). En cas d'hypovolémie, la pente descendante est très raide et la courbe devient horizontale pendant la diastole; la surface sous la courbe est très réduite, signant une absence de flux diastolique. La perfusion d'un vasoconstricteur pour remonter la valeur de la pression artérielle dans ces conditions se traduit par l'apparition d'une bosse sur la partie horizontale de la courbe, qui représente en fait un flux rétrograde totalement inefficace [10].
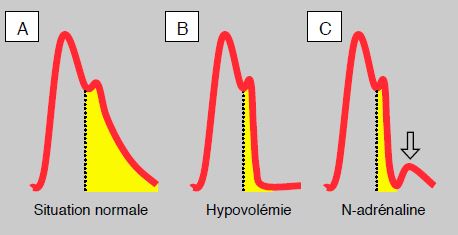
Figure 5.67 : Variation de la surface sous la courbe en diastole. A: situation normale, la pente est oblique et la surface sous la courbe en diastole est importante. B: hypovolémie; la pente est raide et la courbe horizontale pendant la diastole; la surface sous la courbe est minime. C: addition de noradrénaline; il apparaît un petit flux rétrograde (flèche) mais pas d'augmentation de la surface du flux antérograde [d'après référence 7].
La vélocité de l'onde de pression peut se mesurer cliniquement par le délai d'apparition du pouls entre la carotide et la fémorale par tonométrie d'applanation ou par effet Doppler, en prenant l'onde R de l'ECG comme référence temporelle et en mesurant la distance entre le creux sus-sternal et le pli inguinal. De la même manière, l'échocardiographie permet de mesurer le temps écoulé entre la chambre de chasse du VG, l'aorte descendante proximale et l'aorte abdominale; le délai normal est d'environ 40 msec entre chacun des points. La vélocité carotido-fémorale normale est de 7 m/s avant 30 ans, 10 m/s entre 50 et 60 ans et 14 m/s au-delà de 70 ans [6]. Une accélération de plus d'une déviation standard augmente de 50% le risque d'AVC et de maladie cardiovasculaire [3].
Pression artérielle centrale
La pression artérielle centrale est celle qui règne dans l'aorte ascendante (Pao). La pression systolique subit une amplification en allant de l'aorte vers la périphérie parce que les artères sont de moins en moins souples en s'éloignant de l'aorte; la différence sur le pic systolique est en général de 10-20 mmHg mais peut atteindre 40 mmHg, alors que la pression moyenne (PAM) reste stable tout au long de l'arbre vasculaire et que la diastolique diminue un peu [17]. L'accès direct à la pression centrale demande un cathétérisme invasif, mais diverses techniques indirectes permettent de l'évaluer, telles la courbe artérielle par tonométrie carotidienne ou des algorithmes basés la constance de la PAM et la variation de la diastolique. Un enregistrement en haute résolution de la courbe artérielle périphérique permet de différencier un épaulement télésystolique dû à l'onde réfléchie qui correspond en fait à la Pao (Figure 5.68) [20]. On peut aussi considérer que la Pao est égale à 0.9 PAsyst, soit 10% plus basse que la pression artérielle enregistrée au bras [12]. Une autre manière de calcluer est de la considérer comme égale aux deux tiers de la pression pulsée: Pao = (2 PAsyst + PAdiast)/3. La pression centrale est un meilleur prédicteur du risque cardiovasculaire que la pression périphérique, et les anti-hypertenseurs sont efficaces sur le pronostic dans la mesure où ils abaissent la Pao, ce que font les inhibiteurs de l'enzyme de conversion et les bloqueurs calciques mais non les béta-bloqueurs [12].
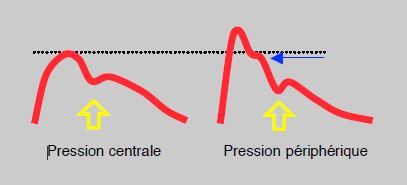
Figure 5.68 : Relation de la pression centrale et de la pression périphérique. Un léger épaulement (flèche bleue) apparaît en télésystole sur la courbe artérielle périphérique; il correspond à la PA systolique de l'aorte ascendante. Flèche jaune: début de la diastole.
Pression pulsée et pulsatilité
Les invertébrés les plus évolués (crabes, poulpes) et l’ensemble des vertébrés disposent d’un ventricule sous-aortique pulsatile comme pompe motrice pour la circulation artérielle; c'est le seul système qui permette de propulser le sang à haute pression. La pulsatilité est une nécessité pour assurer un temps pour le remplissage ventriculaire et pour permettre la perfusion myocardique, puisque le ventricule systémique doit se perfuser lui-même. Seule la diastole garantit un gradient de pression suffisant entre l’aorte et le myocarde pour assurer la perfusion de la paroi ventriculaire, grâce à la présence d'une valve aortique. Même si elle est capitale pour diminuer le risque de thrombose et pour améliorer le flux lymphatique [31], la pulsatilité n’est pas essentielle à la perfusion des organes, comme le prouve l’excellente tolérance à long terme des assistances ventriculaires gauches à flux continu [24]. Cependant, la dépulsation du flux artériel par ces systèmes d'assistance circulatoire à turbine (voir Chapitre 12 Dispositifs à long terme) engendre des modifications vasculaires structurelles dans l'aorte: altération de l'élasticité vasculaire et perte de compliance artérielle, perte cellulaire dans la média avec réaction inflammatoire et dilatation aortique. La conséquence est une rigidité artérielle accrue et une profonde altération du couplage ventriculo-artériel qui rend les malades hypotendus et très sensibles aux variations de volume circulant [14].
De fait, la pulsatilité présente deux inconvénients majeurs du point de vue hydrodynamique.
- Elle représente une augmentation de 15-20% de l’énergie hydraulique totale (PH = Pmoy • Flux) nécessaire par rapport à un flux continu de même débit, puisque la masse sanguine est accélérée et décélérée pendant chaque cycle.
- Elle se prolonge plus ou moins loin en périphérie, alors que le flux capillaire doit être aussi stable que possible pour faciliter les échanges et minimiser le stress de paroi sur l’endothélium et la membrane basale.
C’est pourquoi tout l’arbre artériel est construit pour dépulser au mieux le flux systolo-diastolique éjecté par le ventricule. Par leur expansion en systole, l’aorte et les grandes artères amortissent le pic de pression systolique, baissent la postcharge du VG et emmagasinent de l’énergie qu’elles restituent en diastole sous forme d’une augmentation de pression et de flux. Elles fonctionnent ainsi comme un réservoir-tampon couplé aux artérioles distales à résistance élevée. Cet ensemble permet de fournir un flux quasi-dépulsé dans les capillaires. Pour éviter la dilatation et la rupture, les grandes artères présentent une élasticité non-linéaire : leur compliance est maximale aux dimensions et pressions normales grâce à leur forte teneur en élastine, une macroprotéine très élastique, mais leur rigidité augmente au-delà de ces valeurs à cause de la mise sous tension du collagène qui fonctionne comme une butée. Leur courbe pression/volume a une forme en "J" [23]. Plus on s’éloigne du cœur, moins les artères sont élastiques : il y a deux fois plus d’élastine que de collagène dans l’aorte thoracique, mais deux fois moins dans l’aorte distale. Tous les vertébrés et les invertébrés les plus évolués disposent d’une mécanique vasculaire identique, sauf que la transition entre la zone de pression à haute compliance et la zone de pression élevée où la paroi devient rigide varie selon la pression moyenne des animaux.
Le degré de pulsatilité est fonction de la performance ventriculaire, de la compliance artérielle et des résistances périphériques. L’amortissement de la pulsatilité dans l’aorte disparaît lorsque les vaisseaux sont tendus à cause d’une hypertension artérielle et lorsqu’ils se sclérosent avec l’athéromatose ou se calcifient avec l’âge. Ainsi la pression systolique de la personne hypertendue ou artériosclérotique s’élève parce que ses vaisseaux sont rigides, mais sa diastolique s’abaisse parce qu’il n’y a plus de restitution de pression en diastole : la pression différentielle, ou pression pulsée, augmente. Cette augmentation de la pression pulsée (PP normale : 40-60 mmHg), est directement liée au risque d’accident cardio- et cérébrovasculaire [19]. La mortalité cardiaque postopératoire double lorsque la PP dépasse 80 mmHg. La pression pulsée est l’élément pronostique majeur dans l’hypertension artérielle. En effet, les vaisseaux artériolaires du cerveau, des reins et du cœur ont des résistances plus basses que celles des autres organes et reçoivent un flux encore largement pulsatile ; lorsque la pulsatilité centrale augmente, leurs réseaux microvasculaires n’en sont pas protégés par la vasoconstriction artériolaire systémique, d’où la fragilité de ces organes face à l’hypertension et à l’âge [19]. Le cœur est toutefois moins affecté que le cerveau ou le rein, car ses vaisseaux sont protégés en systole par la contraction myocardique [6].
Entre 20 et 70 ans, l'onde réfléchie double sa vitesse de propagation, ce qui signifie une diminution par quatre de la distensibilité aortique [6]. Cette perte d’élasticité artérielle engendrée par l’âge, l’athéromatose et l’hypertension a quatre conséquences qui sont bien visibles si l'on superpose les courbes de pression enregistrées dans une artère normale et dans une artère rigidifiée (voir Figure 5.66B).
- Augmentation de la pression systolique centrale ; le risque d’accident vasculaire cérébral et la néphropathie hypertensive sont liés à ce phénomène.
- Augmentation de la pulsatilité et de la pression pulsée.
- Augmentation de la surface sous la courbe en systole ; elle élève la postcharge du VG et induit une HVG concentrique.
- Diminution de la surface sous la courbe en diastole ; elle provoque une baisse de la perfusion coronaire diastolique et contribue à l’augmentation de la pulsatilité.
Les personnes âgées sont le plus souvent hypertendues et souffrent en général d’athéromatose aortique. Ces deux phénomènes augmentent l’onde réfléchie et font apparaître un second pic de pression en systole. La vasodilatation liée à l’anesthésie diminue la réflexion en périphérie mais la pression artérielle élevée maintient la vitesse de propagation de l’onde de pression. A l’induction, on voit donc s’amenuiser le pic de l’onde réfléchie sans que sa synchronisation se modifie (Figure 5.69). Comme le moniteur de pression enregistre la valeur maximale comme PAsyst, cette dernière baisse considérablement, alors que la pression due à l’éjection du volume systolique dans le système artériel se modifie dans une moindre mesure. La perfusion des organes ne souffre donc pas de cette chute de la pression systolique réfléchie.
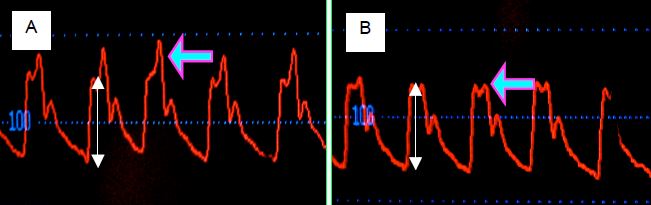
Figure 5.69 : Courbes de pression artérielle avant (A) et après (B) l’induction chez un patient de 80 ans dont les vaisseaux artériels sont athéromateux et rigides. L’induction de l’anesthésie a provoqué une baisse des résistances artérielles périphériques ; cette vasodilatation a diminué considérablement l’intensité de l’onde réfléchie (flèche violette), mais non sa synchronisation parce que la pression artérielle différentielle liée au flux sanguin (premier pic systolique) n’a pas changé (doubles flèches blanches). Le moniteur de pression affiche la valeur maximale de la pression comme valeur systolique ; on note donc une chute importante de la PAsyst, alors que la pression due à l’éjection du volume systolique dans les artères ne s’est presque pas modifiée.
En plus d’être pusatile, le flux dans l’aorte est laminaire et avance avec un mouvement spiralé dû à la torsion du VG sur lui-même pendant la phase d’éjection (voir Figure 5.22). Le sang tournoie en sens anti-horaire [13]. Cette propulsion spiralée présente un double avantage.
- Les forces de cisaillement sont réduites lorsque le sang prend un virage comme la crosse de l’aorte ; si le flux était rectiligne, les couches parallèles auraient des vitesses différentes dans une courbe, celles de l’extérieur devant avancer plus rapidement que celles de l’intérieur.
- La perfusion des vaisseaux de la gerbe (carotides et sous-clavières) est assurée sans perte de charge, car le flux n’a pas à prendre un angle droit par rapport à sa progression dans l’aorte comme ce serait le cas s’il était rectiligne.
Le remodelage du VG dans l’hypertrophie, la dilatation ou la cicatrisation après infarctus altère sa capacité de torsion et perturbe la géométrie du flux aortique ; la performance hémodynamique est altérée, même si les mesures de pression et de débit restent satisfaisantes.
Elastance artérielle
L'élastance artérielle (Ea) et une propriété de l'arbre vasculaire qui évolue de manière inverse à l'élastance ventriculaire au cours du cycle cardiaque. Elle exprime la combinaison de la compliance, de l'impédance et de la résistance des artères; elle englobe l'effet de l'onde réfléchie et de la pulsatilité. Sur une boucle P/V, l’Ea apparaît comme une droite en miroir de l’élastance ventriculaire (Ets ou Emax): l’une augmente lorsque l’autre diminue (Figure 5.70). L'Ea s’élève pendant la systole et rejoint l’Ets au point télésystolique (ts); sa pente est proportionnelle à la résistance artérielle totale [26,27]. Sur le graphique pression/volume, l'Ea est définie comme la droite qui relie le point télésystolique (Pts sur l'axe de la pression) au volume télédiastolique (Vtd sur l'axe du volume). Sa pente est définie par le rapport entre la pression télésystolique et le volume systolique (en mmHg/mL): Ea = Pts/VS [7]. Sa valeur s'étend de 0.4 à 8.4 mmHg/mL [4]. Pour rappel, la pente de l'élastance ventriculaire (Ets ou Emax) est le rapport entre la pression télésystolique et le volume résiduel: Ets = Pts/Vts (on admet que le V0 soit une valeur négligeable). La loi d'Ohm appliquée à l'hémodynamique formule que les résistance artérielles (RAS) sont proportionnelles au rapport entre la pression artérielle et le débit cardiaque: RAS ≈ PAM/DC (Tableau 5.5). La pression est donc proportionnelle au produit du débit cardiaque et des résistances: P ≈ RAS x DC, ou P ≈ RAS x (VS x Fc) où Fc est la fréquence cardiaque. En substituant cette équation dans la formule de l'élastance artérielle réarrangée (Pts = Ea x VS), on obtient: Ea = RAS x Fc. Ainsi l'élastance artérielle est fréquence-dépendante [7]. Comme le travail ventriculaire augmente avec les RAS, le cœur a intérêt à ce que la pente d’Ea soit faible ; il faut toutefois qu’elle représente une résistance suffisante pour maintenir la pression de perfusion adéquate dans l’organisme.
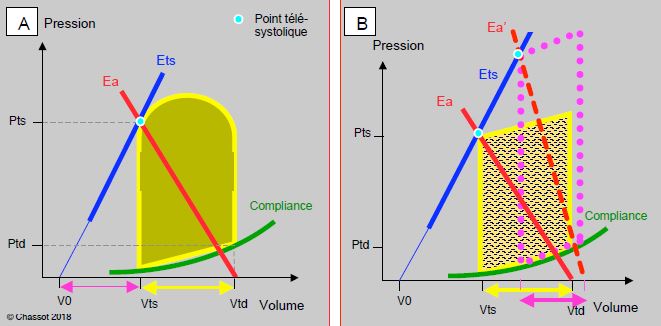
Figure 5.70 : Définition de l’élastance artérielle (Ea) sur un diagramme Pression – Volume. La courbe de l’Ea est une quasi-droite en miroir de l’Emax. Elle est définie par le point télésystolique et le volume télédiastolique sur l'abscisse. Sa pente est légèrement plus faible que celle d’Emax ; de ce fait le volume éjecté (Vtd – Vts, flèche jaune) et un peu plus grand que le volume résiduel (Vts – V0, flèche violette). La fraction d’éjection FE = (Vtd – Vts) / Vtd est donc supérieure à 0.5 (normale : 0.55 - 0.75). La figure de droite représente la valeur de l’élastance artérielle (Ea’) en cas d’hypertension artérielle (boucle pointillée violette); la pente de l’Ea est plus importante, mais le volume systolique plus faible (flèche violette).
L'évaluation de l'Ea est complexe, mais elle est possible de manière non-invasive en combinant différentes mesures: la fraction d'éjection, le volume systolique, les intervalles de temps systoliques et les pressions systolique et diastolique [4]. Plus simplement, elle peut s'exprimer par le rapport: Ea = 0.9 PAsyst/VS, puisque la pression artérielle aortique est environ 10% plus basse que la pression systolique mesurée au bras [11]. Une autre approximation consiste en un renversement de proportion par rapport au calcul de la pression moyenne: Pts = (2 x Psyst + Pdiast)/3 [12]. En ventilation en pression positive, on peut déterminer l'élastance dynamique (Eadyn) qui est le rapport entre la variation de la pression pulsée (ΔPP) et la variation du volume systolique (ΔVS) au cours d'un cycle respiratoire; elle est bien corrélée avec la réponse à l'administration de volume ou de vasopresseur [2,9].
Toutefois, l'élastance traduit mal le fait que la postcharge du VG est une notion pulsatile et dynamique qui se modifie au cours de la systole, puisque le ventricule s'épaissit et se raccourcit; ainsi la tension de paroi est maximale en protosystole et presque de moitié en télésystole (voir Figure 5.49). D'autre part, l'onde de pression réfléchie tend à réaugmenter la postcharge en télésystole. Ainsi, l'élastance ne représente la force qui s'oppose à l'éjection que dans le cadre de la boucle pression/volume [30].

Couplage ventriculo-artériel
L'Ea représente la tension de l'appareil vasculaire alors que l'Ets représente la tension du ventricule. Le couplage ventriculo-artériel (CVA) se définit comme le rapport entre l'élastance artérielle et l'élastance ventriculaire (CVA = Ea/Ets). Lorsqu'il est voisin de 1, toute l'énergie hydraulique du ventricule est transmise au système artériel et la consommation d'énergie est optimale par rapport au travail éjectionnel fourni [26].
La boucle P/V d’un cœur normal montre que la pente d’Ea est un peu plus faible que la pente d’Ets. La valeur normale du rapport Ea/Ets voisine 0.7 pour le VG et 0.5-0.7 pour le VD [1]. C'est la valeur qui fournit le travail hémodynamique au meilleur coût énergétique. Cela a aussi pour effet d’assurer un volume éjecté (Vtd – Vts) plus grand que le volume résiduel (Vts – V0). La fraction d’éjection (rapport entre le volume éjecté et le volume télédiastolique) est donc définie par l’équilibre entre la fonction ventriculaire (Ets) et la fonction artérielle (Ea), et n’est pas en soi un indice de contractilité. Elle exprime au contraire le couplage ventriculo-artériel.
A l'effort, l'Ets augmente davantage que l'Ea, et le rapport Ea/Ets diminue; ceci favorise l'éjection d'un plus grand volume systolique. Avec l'âge, l'Ea augmente et la capacité d'effort diminue puisque le rapport Ea/Ets ne peu plus s'abaisser suffisamment. Le système ventriculo-artériel est découplé lorsque le rapport devient > 1. Dans l'insuffisance cardiaque, le CVA augmente de 3-4 fois parce que l'Ets est très abaissée et l'Ea augmentée [8]. Dans l'hypertension artérielle ou la sénescence, l'Ea est excessivement haute. Dans l'insuffisance diastolique, la fonction systolique est conservée mais le ventricule est rigide, ce qui double l'Ets, déplace vers le haut la courbe de compliance (Ptd élevée) et diminue la capacité à l'effort car la contractilité ne peut pas s'élever davantage lorsqu'elle est sollicitée. La rigidité de l'arbre artériel et du myocarde augmentent la dépense énergétique d'environ 50% par rapport à la normale [11]. L'augmentation simultanée de l'Ets et de l'Ea réalise une situation de couplage serré qui limite considérablement la tolérance aux variations de volume circulant; ces patients sont donc très sensibles aux modifications de précharge, comme à l'induction de l'anesthésie ou lors d'hypovolémie. Dans ces conditions, une baisse de la fréquence cardiaque diminue l'élastance artérielle et soulage le ventricule; c'est un des modes d'action des béta-bloqueurs [7]. A l'effort, le VD réagit comme le VG: l'Ets s'élève plus que l'Ea. Chez les malades souffrant d'hypertension artérielle pulmonaire avancée, par contre, l'Ea augmente davantage que l'Ets, déjà à son maximum, ce qui conduit à un découplage ventriculo-artériel (Ea/Ets > 1) [25]. Cette péjoration du couplage V-A est un prédicteur indépendant de morbi-mortalité chez ces patients [29].
| Couplage ventriculo-artériel |
|
La pression artérielle est le résultat de l'éjection ventriculaire contre les résistances artérielles. Les grandes artères très élastiques (aorte et ses branches) emmagasinent une partie de la pression et du volume d'éjection systolique (abaissement de la PAsyst) et les restituent en diastole (augmentation de la PAdiast). Les artérioles périphériques offrent une grande résistance et dépulsent partiellement le flux. La PAM est identique dans tout l'arbre artériel, alors que la PAsyst augmente en périphérie et que la PAdiast y diminue un peu.
L'onde de pression générée par le ventricule se propage dans l'arbre vasculaire plus vite (4-15 m/s) que le flux artériel (1.0 m/s); elle est réfléchie en périphérie en fonction du degré de vasoconstriction et revient au cœur en protodiastole. Sa vitesse de déplacement augmente lorsque les vaisseaux sont rigides et la pression artérielle élevée; elle se superpose alors à la pression systolique. La rigidité de l'arbre artériel causée par l'athéromatose, l'âge et l'hypertension augmentent la PAsyst et baissent la PAdiast parce que l'amortissement élastique des grandes artères est supprimé; cette augmentation de la pression pulsée (PAsyst – PAdiast) est directement liée aux complications cardio- et cérébrovasculaires.
L'élastance artérielle (Ea) figurée sur la boucle PV est l'équivalent inverse de l'élastance ventriculaire (Ets); sa pente est proportionnelle à la résistance artérielle totale. Le rapport Ea/Ets définit le couplage ventriculo-artériel (valeur normale: 0.7-1.0). La fraction d'éjection est la variable d'ajustement entre la performance systolique du ventricule et la résistance à l'éjection de l'arbre artériel.
|
© CHASSOT PG Août 2010, dernière mise à jour Novembre 2019
Références
- ASANOI H, SASAYAMA S, KAMEYAMA T. Ventriculoarterial coupling in normal and failing heart in humans. Circ Res 1989; 65:483-93
- BAR S, LEVIEL F, ABOU ARAB O, et al. Dynamic arterial elastance measured by uncalibrated pulse contour analysis predicts arterial-pressure response to a decrease in norepinephrine. Br J Anaesth 2018; 121:534-40
- BEN-SHLOMO Y, SPEARS M, BOUSTRED C, et al. Aortic pulse wave velocity improves cardiovascular events predicition: an individual participant meta-analysis of prospective observational data from 17'635 subjects. J Am Coll Cardiol 2014; 63:636-46
- CHEN CH, FETICS B, NEVO E, et al. Noninvasive single-beat determination of left ventricular end-systolic elastance in humans. J Am Coll Cardiol 2001; 38:2028-34
- CHIRINOS JA. SEGERS P, GILLEBERT TC, et al. Arterial properties as determinants of time-varying myocardial stress in humans. Hypertension 2012; 60:64-70
- CHIRINOS JA. SEGERS P, HUGHES T, TOWSEND R. Large-artery stiffness in health and disease. JACC state-of-the-art review. J Am Coll Cardiol 2019; 74:1237-63
- FOX JM, MAURER MS. Ventriculovascular coupling in systolic and diastolic heart failure. Curr Heart Fail Rep 2005; 2:204-11
- GAASCH WH, ZILE MR. Left ventricular structural remodeling in health and disease. J Am Coll Cardiol 2011; 58:1733-40
- GUARRACINO F, BALDASSARRI R, PINSKY MR. Ventriculo-arterial decoupling in acutely altered hemodynamic states. Critical Care 2013; 17:213
- HOSSEINPOUR AR, VAN STEENBERGHE M, BERNATH MA, et al. Improvement in perioperative care in pediatric cardiac surgery by shifting the primary focus of treatment from cardiac output to perfusion pressure: are beta-stimulants still needed ? Cong Heart Dis 2017; 12:570-7
- KAWAGUCHI M, HAY I, FETICS B, KASS DA. Combined ventricular systolic and arterial stiffness in patients with heart failure and preserved ejection fraction. Circulation 2003; 107:714-20
- KELLY RP, TING CT, YANG TM, et al. Effective arterial elastance as index of arterial vascular load in humans. Circulation 1992; 86:513-21
- KILNER PJ, YANG GZ, MOHIADDIN RH, et al. Helical and retrograde secondary flow patterns in the aortic arch studied by three-directional magnetic resonance velocity mapping. Circulation 1993; 88:2235-47
- LEE M, AKASHI H, KATO TS; et al. Vascular inflammation and abnormal aortic histomophometry in patients after pulsatile- and continuous-flow left ventricular assist device placement. J Heart Lung Transpl 2016; 35:1085-91
- LEVICK JR. An introduction to cardiovascular physiology. Oxford: Butterworth-Heinemann, 1995, 116-7
- LEVICK JR. An introduction to cardiovascular physiology. 2nd edition. Oxford, Butterworth-Heinemann, 1995, 255-75
- McENIERY CM, COCKCROFT JR, ROMAN MJ, et al. Central blood pressure: current evidence and clinical importance. Eur Heart J 2014; 35:1719-25
- MURGO JP, WESTERHOF N. Arterial reflections and pressure waveforms in humans. In: YIN FCP, ed. Ventricular/vascular coupling. Clinical, physiological and engineering aspects. New York: Springer Verlag, 1987, 140-58
- O’ROURKE MF, SAFAR ME. Relationship between aortic stiffening and microvascular disease in brain and kidney. Cause and logic of therapy. Hypertension 2005; 46:200-4
- O'ROURKE MF, SEWARD JB. Central arterial pressure and arterial pressure pulse: new views entering the second century after Korotkov. Mayo Clin Proc 2006; 81:1057-48
- O'ROURKE MF, YAGINUMA T, AVOLIO AP. Physiological and pathophysiological implications of ventricular/vascular coupling. Ann Biomed Eng 1984; 12:119-34
- PETTERSEN E, HELLE-VALLE T, EDVARDSEN T, et al. Contraction pattern of the systemic right ventricle. J Am Coll cardiol 2007; 49:2450-6
- SHADWICK RE. Mechanical design in arteries. J Experiment Biol 1999; 202:3305-13
- SLAUGHTER MS, ROGERS JG, MILANO CA, et al. Advanced heart failure treated with continuous-flow left ventricular assist device. N Engl J Med 2009; 361:2241-51
- SPRUIJT OA, DE MAN FS, GROEPENHOF H, et al. The effects of exercise on right ventricular contractility and right ventricular-arterial coupling in pulmonary hypertension. Am J Respir Crit Care Med 2015; 191:1050-7
- SUNAGAWA K, MAUGHAN WL, BURKHOFF D, SAGAWA K. Left ventricular interaction with arterial load studied in isolated canine ventricle. Am J Physiol 1983; 56:586-95
- SUNAGAWA K, SAGAWA K, MAUCHAN WL. Ventricular interaction with the vascular system in terms of pressure-volume relationships. In: YIN FCP, ed. Ventricular/vascular coupling. Clinical, physiological and engineering aspects. New York: Springer Verlag, 1987, 210-39
- THIELE RH, DURIEUX ME. Arterial waveform analysis for the anesthesiologist: past, present, and future concepts. Anesth Analg 2011; 113:766-76
- VANDERPOOL RR, PINSKY MR, NAEIJE R, et al. RV-pulmonary arterial coupling predicts outcome in patients referred for pulmonary hypertension. Heart 2015; 101:37-43
- WEBER T, CHIRINOS JA. Pulsatile arterial haemodynamics in heart failure. Eur Heart J 2018; 39:3847-54
- WRIGHT G. Mechanical simulation of cardiac function by means of pulsatile blood pumps. J Cardiothorac Vasc Anesth 1997; 11:299-309
05 Physiopathologie cardio-vasculaire
- 5.1 Préambule
- 5.2 Couplage de l'excitation et de la contraction myocardiques
- 5.3 La contraction myocardique
- 5.4 Mécanique ventriculaire
- 5.5 Physiopathologie de la systole
- 5.6 Physiopathologie de la diastole
- 5.7 Remplissage veineux
- 5.8 Interactions cardio-respiratoires
- 5.9 Dysfonction ventriculaire gauche
- 5.10 Fonction ventriculaire droite et circulation pulmonaire
- 5.11 Perfusion coronarienne et ischémie myocardique
- 5.12 Conclusions